Định nghĩa:
Bệnh Wilson là bệnh di truyền do sự tích lũy đồng trong cơ thể, chủ yếu là ở gan và não. Nếu không được chẩn đoán và điều trị kịp thời, sự tích lũy đồng có thể gây ra các vấn đề nghiêm trọng, và có thể dẫn đến tử vong. Bệnh Wilson là bệnh di truyền lặn trên nhiễm sắc thể thường đặc trưng bởi sự tích lũy đồng trong cơ thể và các bất thường liên qua đến gan và thần kinh. Đây là một bệnh di truyền hiếm gặp với tỷ lệ mắc là 1/30.000 người. Tuy nhiên, tỷ lệ người mang gen bệnh là 1/100 người. Ở Mỹ, ước tính có khoảng 600 trường hợp mắc bệnh Wilson (tần suất mắc là 1/30.000) và 1% dân số là người mang gen bệnh (tần suất người mang gen bệnh là 1/90).
Bệnh Wilson khởi phát một cách nhanh chóng sẽ dẫn đến các tổn thương nghiêm trọng đến chức năng của gan, encephalopathy, coagulopathy, và thậm chí có thể tử vong nếu không được ghép gan kịp thời.
Bệnh Wilson phổ biến hơn ở nữ hơn so với nam, với tỷ lệ nữ/nam là 4/1. Bệnh khởi phát ở nhiều độ tuổi khác nhau. Với những trẻ bị có các triệu chứng liên quan đến gan thường khởi phát ở độ tuổi từ 10 – 13 và với người trưởng thành có các triệu chứng liên đến thần kinh thường khởi phát ở độ tuổi từ 19 – 20.
Bệnh học:
Đồng là một nhân tố vi lượng, có trong rất nhiều loại thực phẩm. Cơ thể con người chỉ cần một lượng đồng rất nhỏ để duy trì sức khỏe. Ước tính lượng đồng tổng số trong cơ thể là 50–100 mg, trung bình một ngày sẽ nhận khoảng 1- 2 mg. Đồng là một thành phần quan trọng đối nới nhiều loại enzyme, chẳng hạn như enzyme thủy phân oxidase, cytochrome c oxidase, superoxide dismutase và dopamine beta hydroxylase.
Nếu bạn là người thừa hưởng một lỗi di truyền liên quan đến các gen gây bệnh trong bệnh Wilson, cơ thể bạn sẽ không thể đào thải đồng. Bình thường, cơ thể loại bỏ lượng đồng du thừa ra ngoài. Tuy nhiên, với người mắc bệnh Wilson không thể tự đào thải lượng đồng dư thừa, do đó, nó tích tụ lại trong cơ thể, chủ yếu ở gan, não, mắt và thận.
Các vấn đề liên quan đến gan:
40 – 50% trường hợp mắc bệnh Wilson bị rối loạn chức năng gan. Các triệu chứng liên quan đến gan thường xuất hiện đầu tiên. Sự tích lũy đồng gây độc cho gan và do đó thường gây viêm gan, dẫn đến vàng da, đau bụng và buồn nôn. Nếu không được điều trị, sự tổn thương tế bào gan sẽ gây ra xơ gan. Thậm chí, trong trường hợp xơ gan nặng.
Độ tuổi trung bình khởi phát bệnh là 11 tuổi. Hiếm khi các triệu chứng xuất hiện trước 5 tuổi, tuy nhiên cũng có thể chẩn đoán bệnh nhân mắc bệnh Wilson sớm, khoảng 2 tuổi nếu thấy xuất hiện sự tăng hàm lượng các enzyme ở gan.
Các vấn đề liên quan đến não:
40 – 60% trường hợp mắc bệnh Wilson có các dấu hiệu lâm sàng do rối loạn chức năng thần kinh. Đồng tích tụ ở não sẽ gây ra nhiều triệu chứng khác nhau: Các triệu chứng liên quan đến vận động: run tay, di chuyển chậm, không đứng được, khó phát âm, khó nuốt, đau đầu, lên cơn (tái phát-seizure), giãn tĩnh mạch, ban đỏ trong lòng bàn tay, u mạch, một số vấn đề liên quan đến viết. Các triệu chứng thần kinh: suy nhược, không có khả năng tập trung. Người bị bệnh thường hay gây gổ và nhạy cảm và có thể có thay đổi bản tính. Ở typ bệnh này, bệnh phởi phát muộn với độ tuổi trung bình là 18, tuy nhiên cũng có thể các triệu chứng thần kinh xuất hiện trước 6 tuổi. Nếu không được điều trị, sự tích lũy đồng ở não có thể gây ra các vấn đề nghiêm trọng chẳng hạn như yếu cơ nặng, mất trí, rigidity.
Các vấn đề khác:
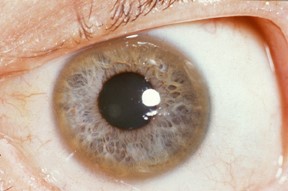
Đồng có thể tích tụ ở mắt với đặc điểm đặc trưng là giác mạc có đặc điểm được gọi là Kayser – Fleischer ring – một màng màu nâu ở giác mạc mắt. Một số dấu hiệu khác có thể xuất hiện bao gồm: thiếu máu, tổn thương thận, vấn đề tim mạch, viêm tụy, các vấn đề bất thường về kinh nguyệt và liên tục xảy thai ở phụ nữ.
Cơ sở phân tử:
Bệnh Wilson do đột biến trên gen ATP7B (WD) nằm trên nhiễm sắc thể 13 (13q14-q21) và có chức năng vận chuyển đồng trong cơ thể. Gen WD gồm 21 exon, mã hóa cho 1411 axit amin, chủ yếu biểu hiện ở gan, thận, placenta. Gen mã hóa cho enzyme copper-transporting P-type ATPase (ATP7B).
Có khoảng 400 đột biến đã được tìm thấy trên gen ATP7B, bao gồm: đột biến mất đoạn nhỏ và đột biến thêm một hoặc một vài cặp base (31%); nonsence mutation (8%); splice site mutation (7%); missense mutation (54%). Hầu hết bệnh nhân Wilson mang hai loại đột biến khác nhau trên cả hai allen nằm trên nhiễm sắc thể. Trong đó đột biến missence là phổ biến nhất. Hầu hết trường hợp bị bệnh là dị hợp hai loại đột biến.
Một số loại đột biến thường gặp như:
- H1069Q (missence mutation): đây là đột biến phổ biến nhất tại Mỹ và phía bắc Châu Âu.
- L936 X (nonsence mutation)
- Q 289X (framshift mutation)
- 2530del A (deletion mutation)
Ở người mắc bệnh Wilson, một phần gen nằm trên nhiễm sắc thể 13 không hoạt động. Gen ATP7B kiểm soát con đường đào thải đồng dư thừa ra khỏi tế bào gan. Bình thường, tiết lượng đồng dư thừa vào mật. Nhưng, nếu quá trình này không hoạt động, đồng sẽ tích tụ trong gan và đến một lượng nhất định sẽ làm cho tế bào gan không thể hoạt động được, lượng đồng dư thừa này sẽ tràn vào máu và tích lại ở khác phần khác của cơ thể, chủ yếu là ở não.
Cơ chế di truyền:
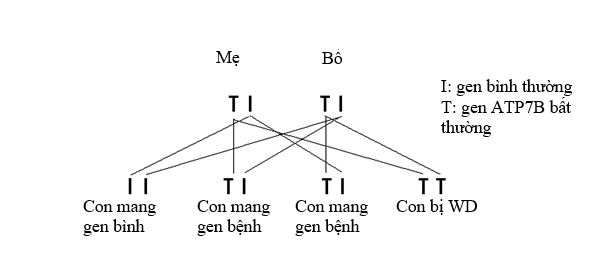
Bệnh Wilson là di truyền lặn trên nhiễm sắc thể thường. Nghĩa là để có thể biểu hiện ra bệnh, bạn phải được thừa hưởng hai gen ATP7B bất thường, một gen từ bố và một gen từ mẹ. Nếu bạn chỉ mang duy nhất một gen bất thường, bạn được gọi là người mang gen bệnh. Người mang gen bệnh không phải là người mắc bệnh di truyền vì họ còn có một gen bình thường để kiểm soát hoạt động chức năng của đồng trong cơ thể. Nhưng người mang gen bệnh có thể truyền lại gen bất thường này cho con cháu họ. Khi hai người cùng mang một gen bất thường, các trường hợp mà con họ sinh ra có thể gặp là:
- 25% đứa trẻ bị bệnh Wilson vì nhận cả hai gen bất thường từ bố và mẹ
- 50% đứa trẻ không bị bệnh Wilson nhưng sẽ là người mang gen bệnh vì nhận một gen bất thường từ bố hoặc mẹ và một gen bình thường từ người còn lại.
- 25% đứa trẻ không bị bệnh Wilson và sẽ không phải là người mang gen bệnh vì nhận cả hai gen bình thường từ bố và mẹ.
Chẩn đoán xác đinh: Bệnh Wilson được chẩn đoán bằng các xét nghiệm sau:
- Xét nghiệm máu để đo hàm lượng ceruloplasmin: đây là một trong những xét nghiệm sàng lọc bởi nó không đặc hiệu cho bệnh Wilson. 5-10% trường hợp mắc bệnh Wilson có lượng ceruloplasmin giảm nhẹ, 10-20% trường dị hợp cũng có lượng ceruloplasmin giảm sút. Sự bất thường về hàm lượng ceruloplasmin có thể do các nguyên nhân khác như viêm gan mãn tính, viêm ruột loét miệng.
- Xét nghiệm nước tiểu để đo hàm lượng đồng: lượng đồng trong nước tiểu của trường hợp mắc bênh Wilson vượt quá 100 µg/d. Tuy nhiên, sự bất thường này cũng có thể do các nguyên nhân khác ví dụ như bệnh gan obstructive. Ở người mang gen bệnh, hàm lượng đồng tăng cao, nhưng không vượt quá ngưỡng 100 µg/d.
- Kiểm tra mắt: Trường hợp có các dấu hiệu liên quan đến rối loạn thần kinh và tâm thần thường xuất hiện Kayser -Fleischer ring, nhưng lại không có ở trường hợp rối loạn chức năng gan.
- Sinh thiết gan: Sinh thiết gan để xác định hàm lượng đồng là một trong những xét nghiệm nhạy và chính xác để chẩn đoán xác định người mắc bệnh Wilson. Ở người bình thường, lượng đồng từ 15 – 55 µg/g. 83,3% bệnh nhân mắc bệnh Wilson có lượng đồng trong cơ thể cao hơn 250 µg/g, 3,5% còn lại thấp hơn 50 µg/g. Tuy nhiên, tăng lượng đồng trong gan có thể do các bệnh về gan chẳng hạn như viêm gan mãn tính, cirrhosis… Tuy nhiên, sinh thiết gan chỉ được sử dụng trước hết cho bệnh nhân rối loạn chức năng gan mà không cần thiết đối với những trường hợp có các dấu hiệu rối loạn thần kinh.
- Chẩn đoán hình ảnh: 100% bệnh nhân mắc bệnh Wilson mà có các biểu rối loạn thần kinh hầu như đều có bất thường khi tiến hành MRI với các đặc điểm là giãn não thất, teo vỏ não và một số bất thường khác.
Phương pháp xét nghiệm: Phân tích tìm đột biến trên gen ATP7B
- Tách chiết DNA
- PCR tìm đột biến trên từng exon
- SSCP (Single strange conformational polymorphism) tìm đột biến trên vùng không phiên mã 5’-UTR
- Giải trình tự toàn bộ gen ATP7B
TÀI LIỆU THAM KHẢO
Leggio L, Malandrino N, Loudianos G, Abenavoli L, Lepori MB, Cappristo E, Virgiliis SD, Gasbarrini G, Addolorato G (2007), Analysis of the T1288R mutation of the Wilson disease ATP7B gene in four generations of a family: Possible genotype-phenotype correlation with hepatic onset, Dig. Dis. Sci, 52. pp. 2570-2575.
Cox DW, Moore SDP (2002), Copper transporting P-type ATPase and human disease, Journal of Bionergenetics and Biomembrane, 34, pp.5.
Shah R, Piper MH (2009), Wilson disease, emedicine. published online at http://emedicine.medscape.com/article/183456-diagnosis
Ronald F, Pfeiffer (2007), Wilson ‘s disease, Semin Neuro, 27.2, pp. 123-132.