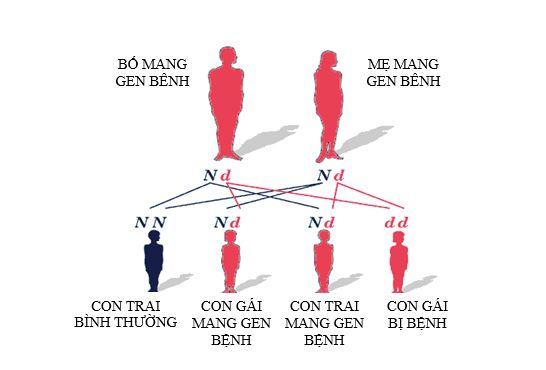
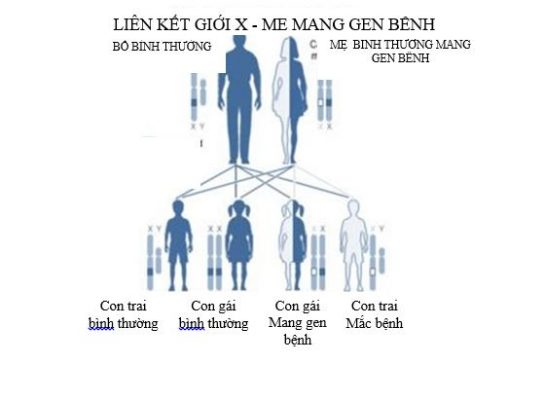
Teo cơ tuỷ (Spinal muscular atrophy – SMA) là bệnh thần kinh cơ, di truyền lặn do đột biến gene trên nhiễm sắc thể thường. Tỷ lệ mắc bệnh là 1/10.000 trẻ đẻ sống và tỷ lệ người mang gene bệnh là 1/50.
Bệnh SMA đặc trưng bởi sự suy yếu của các cơ gốc chi đối xứng do thoái hoá tuần tiến của các tế bào sừng trước tuỷ sống. SMA được chia thành ba thể lâm sàng dựa vào tuổi khởi phát bệnh và mức độ nặng của bệnh: thể nặng, thể vừa và thể nhẹ tương đương với SMA type I, II và III.
Bệnh loạn dưỡng cơ Duchenne (DMD) là một trong những bệnh thần kinh cơ – di truyền phổ biến nhất với tỷ lệ mới mắc là 1/3.500 trẻ trai đẻ sống.
Bệnh DMD có đặc trưng là thoái hoá và gây suy yếu cơ một cách tuần tiến dẫn đến tàn tật và tử vong do suy tim và bội nhiễm phổi. Các dấu hiệu lâm sàng của bệnh được nhận biết ở giai đoạn trẻ từ 2 đến 3 tuổi. Các bệnh nhân được coi như mắc thể nặng nếu phải ngồi xe lăn, mất khả năng tự đi lại trước tuổi 12.
Tăng sản thượng thận bẩm sinh (TSTTBS) là bệnh di truyền lặn trên nhiễm sắc thể thường, được đặc trưng bởi sự thiếu hụt hoạt động của một trong các enzyme cần thiết để tổng hợp cortisol từ cholesterol của vỏ thượng thận như: 21-hydroxylase (21-OH), 11β-hydroxylase (11-OH), 17- hydroxylase (17-OH), 3β- hydroxysteroid dehydrogenase (3β- HSD) và 20/22 Desmolase. Trong đó, 90- 95% trường hợp TSTTBS là do thiếu hụt hoạt động của enzyme 21-OH.
Thiếu hụt citrin (Citrin deficiency) là bệnh di truyền lặn trên nhiễm sắc thể thường, do đột biến gen SLC25A13 nằm trên cánh dài NST số 7 qui định (7q21.3). Citrin – protein vận chuyển Aspartate-Glutamate (Aspartate Glutamate Carrier), đóng vai trò thiết yếu trong quá trình tổng hợp ure và kênh vận chuyển aspartate-malate (aspartate-malate shuttle-MA shuttle). Sự thiếu hụt Citrin ảnh hưởng đến quá trình tổng hợp protein, tái tạo glucose và sinh tổng hợp nucleotit.
Ở khu vực Đông Á, tần suất người mang gen bệnh trong các quần thể không đồng đều: ở Nhật Bản: 1/65; Trung Quốc: 1/63; Hàn Quốc: 1/108. Hiện chưa có nghiên cứu nào về tần suất người mang gen bệnh tại Việt Nam.
Beta thalassemia là bệnh di truyền lặn nhiễm sắc thể thường do đột biến gen β-globin nằm trên cánh ngắn NST 11 qui định (11p15.5), gây giảm hoặc mất tổng hợp chuỗi β globin.
Beta thalassemia là một trong những bệnh huyết sắc tố phổ biến nhất, phân bố khắp thế giới. Ở Việt Nam, theo Nguyễn Công Khanh, bệnh β thalassemia là nguyên nhân hàng đầu gây thiếu máu, tan máu nặng ở trẻ em.
Tỷ lệ người mang gen bệnh phân bố trong cả nước và khác nhau tùy từng địa phương, từng nhóm dân tộc. Đặc biệt, tỷ lệ mang gen bệnh rất cao ở các dân tộc ít người như: Mông (25%), Catu (14%), Tày (11%), Pako (8.33%).
Alpha thalassemia là một trong các bệnh di truyền lặn nhiễm sắc thể thường phổ biến nhất thế giới. Bệnh do đột biến gen HbA, nằm trên cánh ngắn nhiễm sắc thể 16 (16p13.3) gây ra sự giảm hoặc mất tổng hợp chuỗi alpha globin.
Hemoglobin ở người trưởng thành bình thường có 2 chuỗi alpha globin và 2 chuỗi beta globin. Chuỗi alpha globin được tổng hợp từ 4 gen, trong đó có 2 gen HbA1 và 2 gen HbA2. Số lượng của alpha globin phụ thuộc vào số gene hoạt động. Người càng có ít gene hoạt động thì càng mắc thể bệnh alpha thalassemia nặng hơn.
Yếu tố biệt hóa tinh hoàn (Testis determining factor – TDF) là khái niệm chung về một gen hoặc về sản phẩm của gen đó, biểu hiện đặc điểm của người nam.
U nguyên bào thần kinh (Neuroblastoma), là khối u thể rắn ngoài não thường gặp nhất ở trẻ em. Đây là loại u phôi thai của các tế bào thần kinh ngoại bì có nguồn gốc từ mào thần kinh và hướng đến tủy thượng thận cũng như hệ thần kinh giao cảm. U nguyên bào thần kinh chiếm 5 – 7% số ca ung thư ở trẻ em, tỷ lệ mắc 1/6000 trẻ nhỏ hơn 5 tuổi trong đó tuổi trung bình là 2 và khoảng 25% số ca mới là dưới 1 tuổi.
Để chẩn đoán và tiên lượng bệnh, cùng với các thông số về giai đoạn, tuổi bệnh nhân, giải phẫu bệnh, các thay đổi về di truyền như: khuyếch đại gen NMYC, mất đoạn 1p và thêm đoạn 17q cũng là một trong các chỉ thị tiên lượng quan trọng.
Hội chứng thực bào máu – Hemophagocytic lymphohistiocytosis (HLH) là hội chứng trong đó có sự nhân rộng của các tế bào lympho và các đại thực bào trưởng thành, trong một số trường hợp có kèm theo hiện tượng thực bào máu, xuất hiện trước tiên ở lách, hạch lympho, tủy xương, gan, dịch não tủy.
HLH có thể xuất hiện ngẫu nhiên trong quần thể (HLH thứ phát), hoặc có thể do yếu tố gia đình, thuộc loại di truyền lặn trên nhiễm sắc thể thường (Familial Hemophagocytic Lymphohistiocytosis – FHL). Tần suất của FHL vào khoảng 1:50.000 trẻ đẻ sống.
Tuổi xuất hiện bệnh của FHL gặp điển hình ở trẻ nhũ nhi hoặc trẻ nhỏ, tuy nhiên đôi khi cũng gặp ở trẻ lớn hơn. Thông thường, trẻ mắc FHL nếu không được điều trị bằng ghép tế bào gốc tạo máu, sẽ tử vong. Cả hai nhóm HLH thứ phát và FHL đều có thể khởi phát sau quá trình nhiễm khuẩn, chủ yếu là Ebstein Bar Virus (EBV), nên rất khó có thể phân biệt hai nhóm này bằng các dấu hiệu lâm sàng, nếu không sử dụng các phương pháp xét nghiệm sinh học phân tử.
Chẩn đoán bệnh X-linked Lymphoproliferative (XLP) nên được đặt ra ở những trường hợp người nam với biểu hiện nhiễm cấp Epstein-Barr virus (EBV), làm tăng bạch cầu đơn nhân trung tính, có thể kèm theo hiện tượng thực bào tế bào máu. Ở các trường hợp này, nếu bệnh nhân vượt qua được giai đoạn nhiễm EBV tiên phát, có sự biểu hiện của suy giảm chỉ số gammaglobulin máu ở các mức độ khác nhau, và có nguy cơ cao với việc phát triển bệnh thành u lumpho ác tính hoặc các nhóm bệnh có tăng sinh tế bào lympho khác. Các triệu chứng của bệnh XLP đôi khi cũng xuất hiện trên một số bệnh nhân không nhiễm EBV.
Bệnh Wilson là bệnh di truyền do sự tích lũy đồng trong cơ thể, chủ yếu là ở gan và não. Nếu không được chẩn đoán và điều trị kịp thời, sự tích lũy đồng có thể gây ra các vấn đề nghiêm trọng, và có thể dẫn đến tử vong. Bệnh Wilson là bệnh di truyền lặn trên nhiễm sắc thể thường đặc trưng bởi sự tích lũy đồng trong cơ thể và các bất thường liên qua đến gan và thần kinh. Đây là một bệnh di truyền hiếm gặp với tỷ lệ mắc là 1/30.000 người. Tuy nhiên, tỷ lệ người mang gen bệnh là 1/100 người. Ở Mỹ, ước tính có khoảng 600 trường hợp mắc bệnh Wilson (tần suất mắc là 1/30.000) và 1% dân số là người mang gen bệnh (tần suất người mang gen bệnh là 1/90).
Bệnh Wilson khởi phát một cách nhanh chóng sẽ dẫn đến các tổn thương nghiêm trọng đến chức năng của gan, encephalopathy, coagulopathy, và thậm chí có thể tử vong nếu không được ghép gan kịp thời.
Bệnh Wilson phổ biến hơn ở nữ hơn so với nam, với tỷ lệ nữ/nam là 4/1. Bệnh khởi phát ở nhiều độ tuổi khác nhau. Với những trẻ bị có các triệu chứng liên quan đến gan thường khởi phát ở độ tuổi từ 10 – 13 và với người trưởng thành có các triệu chứng liên đến thần kinh thường khởi phát ở độ tuổi từ 19 – 20.
Chứng tạo sinh xương bất toàn hoặc hội chứng xương bất toàn (Osteogenesis imperfecta) – OI, tức là hệ xương không được hình thành một cách hoàn hảo như bình thường, do sự khuyết lỗi của bộ máy di truyền trong vai trò quy định khả năng chịu lực của xương. Do vậy ở những người mắc bệnh này, xương không có độ bền dẻo, mà lại giòn và vì thế xương rất dễ biến dạng và gãy ngay cả khi không có sang chấn hoặc sang chấn rất nhẹ như ho, hắt hơi, vỗ vai….Cũng vì thế mà còn gọi nôm na là “xương thuỷ tinh” để chỉ bệnh này.
Tỷ lệ mắc bệnh vào khoảng 1/10.000 hoặc 1/20.000 trẻ đẻ sống. Tại Mỹ, hiện nay có khoảng 20.000 đến 50.000 người mắc chứng bệnh OI. Tỷ lệ mắc hàng năm của chứng bệnh này ở Mỹ vào khoảng 0.01% .