Định nghĩa:
Bệnh loạn dưỡng cơ Duchenne (DMD) là một trong những bệnh thần kinh cơ – di truyền phổ biến nhất với tỷ lệ mới mắc là 1/3.500 trẻ trai đẻ sống.
Bệnh DMD có đặc trưng là thoái hoá và gây suy yếu cơ một cách tuần tiến dẫn đến tàn tật và tử vong do suy tim và bội nhiễm phổi. Các dấu hiệu lâm sàng của bệnh được nhận biết ở giai đoạn trẻ từ 2 đến 3 tuổi. Các bệnh nhân được coi như mắc thể nặng nếu phải ngồi xe lăn, mất khả năng tự đi lại trước tuổi 12.
Cơ sở phân tử:
DMD là bệnh di truyền lặn liên kết giới tính, có bản chất là do đột biến gene Dystrophin nằm trên cánh ngắn của Nhiễm sắc thể giới tính X (Xp21). Gene Dystrophin có kích thước 2500 Kb gồm ít nhất 79 exon. Sản phẩm của gene Dystrophin là mRNA có kích thước 14 Kb và mã hóa cho protein Dystrophin có kích thước 427 kDa, chức năng của Dystrophin chưa hoàn toàn sáng tỏ nhưng được giả thiết là có tác dụng giữ cho màng sợi cơ được vững chắc.
Các đột biến có thể xảy ra trên gene là đột biến mất đoạn, đột biến điểm, chuyển đoạn và mất đoạn nhỏ. Mất đoạn là loại đột biến hay gặp nhất, chiếm khoảng 65% trong số các đột biến. Trong đó, đột biến mất đoạn hay xảy ra trong vùng “hot spots” của gen, chủ yếu từ exon 44 đến exon 52 và từ exon 2 đến exon 19. Nhận biết đột biến mất đoạn của 25 exon đặc hiệu trong vùng “hotspot” có thể phát hiện được 98 % trong tổng số đột biến mất đoạn của gene Dystrophin.
Việc chẩn đoán chính xác bệnh DMD có tầm quan trọng rất lớn, là tiền đề và cơ sở cho việc phòng bệnh chủ động bằng tư vấn di truyền, chẩn đoán trước sinh cho gia đình người bệnh và điều trị bằng liệu pháp gen trong tương lai.
Triệu chứng lâm sàng:
Tiêu chuẩn cổ điển của Duchenne đã mô tả bao gồm: phả hệ di truyền có người trong gia đình mắc bệnh; trẻ trai; giảm vận động, yếu cơ gốc chi (dấu hiệu Gowers), teo cơ gốc chi nhiều vị trí, đối xứng hai bên, bắp chân phì đại đối xứng hai bên, điện cơ đồ có hình ảnh tổn thương nguồn gốc sợi cơ; men creatine kinase huyết thanh tăng cao trên 20 lần so với trị số bình thường. Các giai đoạn tiến triển của bệnh được chia theo các mức độ sau:
- Giai đoạn nhẹ: trẻ mệt mỏi, và/hoặc bất kỳ biểu hiện yếu cơ nào được phát hiện bao gồm khó di chuyển, dễ ngã, dáng đi bất thường, đi nhón chân, chạy chậm và không có dấu hiệu Gowers.
- Giai đoạn trung bình: dấu hiệu Gowers dương tính, trèo bậc thang khó khăn và/hoặc dáng đi lạch bạch.
- Giai đoạn nặng: không có khả năng tự đứng dậy được nếu không có sự trợ giúp và/ hoặc chỉ bước đi được khi có sự gắng sức, và/ hoặc teo cơ nặng.
- Giai đoạn cuối: phải ngồi xe lăn.
Cơ chế di truyền:
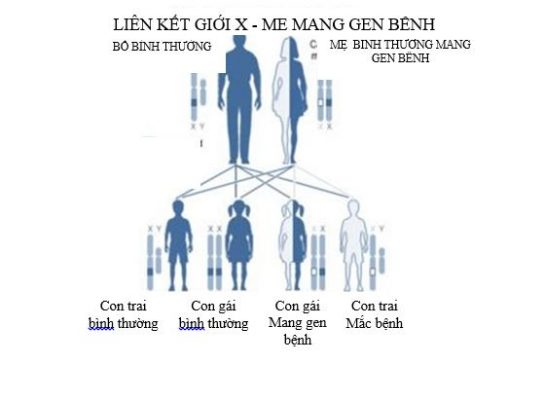
Lọan dưỡng cơ Duchenne là bệnh di truyền liên kết nhiễm sắc thể giới tính X. Mẹ là người bình thường mang gen bệnh, di truyền bệnh cho con trai. Trong một lần sinh, nguy cơ của những người mẹ này như sau:
- 50% số con gái là người bình thường mang gene bệnh.
- 50% số con gái là người bình thường không mang gen bệnh
- 50% số con trai là người bình thường không mang gen bệnh
- 50% số con trai là người mắc bệnh Lọan dưỡng cơ Duchenne
Chẩn đoán xác định:
- Triệu chứng lâm sàng
- Phân tích gene Dystrophin
Phương pháp xét nghiệm:
Phân tích 25 exon vùng hotspot trên gen Dystrophin bằng kỹ thuật Multiplex PCR
- Chiết tách DNA
- Kỹ thuật Multiplex PCR: Kỹ thuật Multiplex PCR thay thế cho kỹ thuật PCR cổ điển để nhận biết sự mất đoạn của 25 exon đặc hiệu tập trung ở vùng “hotspots” của gen. 25 exon này được chia thành 3 bộ Multiplex A, B, C. Sản phẩm PCR được điện di trên gel và phân tích kết quả.
Bảng 1. Các exon của Multiplex A, B, C
| Multiplex A | Multiplex B | Mutiplex C | |||
| Exon | Kích thước (bp) | Exon | Kích thước (bp) | Exon | Kích thước (bp) |
| 45 | 547 | Pm | 535 | 49 | 439 |
| 48 | 506 | 3 | 410 | Pb | 332 |
| 19 | 459 | 43 | 357 | 16 | 290 |
| 17 | 416 | 50 | 271 | 41 | 270 |
| 51 | 388 | 13 | 238 | 32 | 253 |
| 8 | 360 | 6 | 202 | 42 | 195 |
| 12 | 331 | 47 | 181 | 34 | 171 |
| 44 | 268 | 60 | 139 | ||
| 4 | 196 | 52 | 113 | ||
Phân tích toàn bộ 79 exons trên gen Dystrophin bằng kỹ thuật MLPA (Multiplex Ligation – Dependent Probe Amplification):
- Chiết tách DNA
- Kỹ thuật MLPA: Kit Salsa MLPA do MRC-Holland, Amsterdam Hà Lan sản xuất, chứa 80 đoạn dò cho 79 exon đích và exon DP427c. 80 probe này chia đều vào hai hỗn hợp probe P034A2 và P035A2. 50-200ng DNA của bệnh nhân được biến tính ở 980Cx5’, làm lạnh trên đá rồi trộn với hỗn hợp probe và MLPA buffer. Hỗn hợp DNA và Probe được ủ lai ở 950Cx15’ và 600Cx16h hoặc qua đêm. Phản ứng nối ghép sau lai được thực hiện với hốn hợp Ligase-65 và buffer. Sản phẩm nối ghép dược trộn với PCR buffer và mastermix. Sau phản ứng PCR, sản phẩm khuyếch đại được điện di trên hệ thống CEQ8000.
- Phân tích kết quả: Kết quả được biểu thị dưới dạng biểu đồ sóng và dưới dạng bảng. Các vị trí gen bị mất đoạn ở cá thể nam (1 nhiễm sắc thể X) sẽ không có đỉnh tín hiệu xuất hiện.
Tài liệu tham khảo:
Kunkel, L. M. et al. 1991. Searching for dystrophin gene deletion in patients with atypical presentations; in Etiology of human disease at the DNA level. Ravel, New York. pp 51 – 60.
Roberts R G, Bobrow M, Bentley D R. 1992. Point mutations in the dystrophin gene. Proc Natl Acad Sci USA. 89(6): 2331 – 2335.Roberts R G, Coffey A J, Bobrow M, Bentley D R. 1993. Exon structure of the human dystrophin gene. Genomics. 51(6): 919 – 928.
Thomas W. Prior, Scott J. Bridgeman. 2005. Experience and Strategy for the Molecular testing of Duchenne Muscular Dytrophy. Journal of Molecular Diagnostics. Vol. 7, No. 3.
Yuge L., Hui L., Bingdi X. 1999. Detection of gene deletions in Chinese patients with Duchenne/ Becker muscular dystrophy using cDNA probes and the polymerase chain reaction method. Lif Sci. 65: 863 – 869.