Định nghĩa:
Teo cơ tuỷ (Spinal muscular atrophy – SMA) là bệnh thần kinh cơ, di truyền lặn do đột biến gene trên nhiễm sắc thể thường. Tỷ lệ mắc bệnh là 1/10.000 trẻ đẻ sống và tỷ lệ người mang gene bệnh là 1/50.
Bệnh SMA đặc trưng bởi sự suy yếu của các cơ gốc chi đối xứng do thoái hoá tuần tiến của các tế bào sừng trước tuỷ sống. SMA được chia thành ba thể lâm sàng dựa vào tuổi khởi phát bệnh và mức độ nặng của bệnh: thể nặng, thể vừa và thể nhẹ tương đương với SMA type I, II và III.
Triệu chứng lâm sàng:
Loại trừ các bệnh nhân có biểu hiện giảm trương lực cơ toàn thân do các nguyên nhân khác: tổn thương não, bất thường nhiễm sắc thể (hội chứng Prader Willi…)…
- SMA type I (Bệnh Werdnig-Hoffmann, OMIM 253300): Bệnh xuất hiện trước 6 tháng tuổi. Trẻ có biểu hiện khó thở, khó nuốt, giảm trương lực cơ toàn thân nặng, mất phản xạ gân xương, co rút cơ cục bộ (lưỡi, mặt), không tự ngồi được. Trẻ phát triển tinh thần bình thường.
- SMA type II (OMIM 253550): Bệnh xuất hiện trước 18 tháng tuổi. Trẻ có biểu hiện giảm trương lực cơ, chậm phát triển vận động, có thể ngồi được nhưng không đứng và đi được. Có biến chứng cong vẹo cột sống và nuốt khó. Trẻ phát triển tinh thần bình thường.
- SMA type III (Bệnh Kugelberg-Welander, OMIM 253400): Bệnh xuất hiện sau 2 tuổi. Trẻ chậm phát triển vận động, tự đi được nhưng muộn. Có các biểu hiện của yếu cơ gốc chi, co rút cơ, cong vẹo cột sống. Trẻ phát triển tinh thần bình thường.
Cơ sở phân tử:
Vùng gene gây bệnh của cả ba thể lâm sàng nằm trên cánh dài của nhiễm sắc thể số 5 (5q13), có kích thước 750 Kb và có cấu trúc phức tạp. Vùng gene này có cấu trúc lặp và đảo ngược đối xứng với ít nhất ba gene ở phía cuối (Telomeric – T) và bản sao ở vùng trung tâm (Centromeric – C): gene SMN (survival motor neuron); gene NAIP (neuronal apoptosis inhibitory protein) và gene P44.
Gene SMN bao gồm 9 exon: exon 1, 2a, 2b và exon 3 – 8. Gene SMN có 2 bản sao giống nhau là gene SMN-T (SMN 1) và gene SMN-C (SMN 2). Hai gene này chỉ khác nhau ở 5 cặp base: một cặp ở intron 6, một cặp ở exon 7, hai cặp ở intron 7 và một cặp ở exon 8. Sự khác nhau ở exon 7 và exon 8 của hai gene này được sử dụng để phân biệt giữa gene SMN-T và SMN-C và trong chẩn đoán SMA . Qua các nghiên cứu cho thấy, ở 96,4% các bệnh nhân SMA có sự mất đoạn đồng hợp tử exon 7 của gene SMN-T. Vì vậy có thể khẳng định rằng đột biến mất đoạn của gene SMN-T là nguyên nhân chủ yếu gây ra SMA. Sự mất đoạn của gene SMN-C cũng được phát hiện ở 5-10% người không có biểu hiện lâm sàng của SMA, vì vậy mất đoạn gene SMN-C không biểu hiện ra kiểu hình.
Trong thực hành lâm sàng nhi khoa, việc chẩn đoán nguyên nhân của các trường hợp yếu cơ gốc chi nguồn gốc tổn thương thần kinh ngoại vi thường rất phức tạp, đặc biệt là trong trường hợp giảm trương lực cơ ở trẻ nhũ nhi (floppy infants). Nguyên nhân gây ra bệnh cảnh lâm sàng “floppy child” rất phức tạp và đa dạng trong đó có SMA. Do đó việc phát hiện đột biến ở bệnh nhân có triệu chứng nghi ngờ SMA là rất cần thiết. Đây là bước khởi đầu cho tư vấn di truyền và chẩn đoán trước sinh.
Cơ chế di truyền:
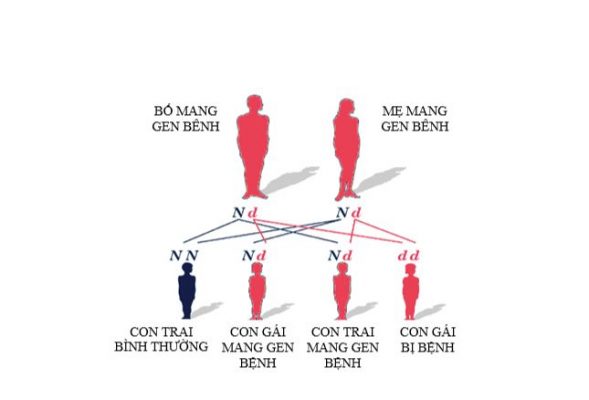
Thoái hóa cơ tủy (SMA) là bệnh di truyền lặn trên NST thường. Tỷ lệ mắc bệnh là như nhau ở cả hai giới nam và nữ.
Bố và mẹ là mang gen dị hợp tử với một đột biến, nguy cơ trong một lần sinh như sau:
- 50% số con là người lành mang gen bệnh.
- 25% số con là người hoàn toàn khỏe mạnh không mang gen bệnh
- 25% số con mắc bệnh SMA.
Chẩn đoán xác định:
- Triệu chứng lâm sàng
- Dựa vào phân tích gene SMN-T và SMN-C
Phương pháp xét nghiệm: Phân tích gene SMN-T và gene SMN-C:
- Tách chiết DNA
- Kỹ thuật PCR: Trình tự mồi cho exon 7 của gene SMN được thiết kế theo Vander Steege và cs (R111 và X7-Dra). Sản phẩm PCR được kiểm tra bằng quá trình điện di trên agarose 1%. Sản phẩm PCR có kích thước 194bp.
- Cắt enzyme: Sản phẩm PCR được tiến hành cắt enzyme để nhận biết exon 7 của gene SMN-T và SMN-C.
- Điện di sản phẩm: Tiến hành quá trình điện di trên Agarose để nhận biết kích thước của các exon. Sản phẩm sau khi cắt enzyme là exon 7 của gene SMN-T (194bp) và exon 7 của gene SMN-C (171bp + 23bp). Mất đoạn đồng hợp tử exon 7 của gen SMN-T (194bp) gây bệnh thoái hóa cơ tủy.
Tài liệu tham khảo:
Ogino S., Hanna Rennert and Robert B. Wilson. 2002. Spinal muscular atrophy genetic testing experience at an Academic medical center. J Mol Diagn. 4(1):8– 53.
Scheffer H., Cobben J. M., Matthijs G., Wirth B. 2001. Best Practice Guidelines for Molecular Analysis in Spinal Muscular Atrophy. European Molecular Genetics Quality Network (EMQN).
Wirth B., Herz M., Wetter A., Moskau S., Hahnen E., Rudnik – Schoneborn, Wienker T., Zerres K. 1999. Quantitative analysis of survival motor neuron copies: Indentification of subtle SMN 1 mutaions in patients with spinal muscular atrophy genotype, phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 64: 1340–1356.