Thiếu hụt citrin (Citrin deficiency) là bệnh di truyền lặn trên nhiễm sắc thể thường, do đột biến gen SLC25A13 nằm trên cánh dài NST số 7 qui định (7q21.3). Citrin – protein vận chuyển Aspartate-Glutamate (Aspartate Glutamate Carrier), đóng vai trò thiết yếu trong quá trình tổng hợp ure và kênh vận chuyển aspartate-malate (aspartate-malate shuttle-MA shuttle). Sự thiếu hụt Citrin ảnh hưởng đến quá trình tổng hợp protein, tái tạo glucose và sinh tổng hợp nucleotit.
Ở khu vực Đông Á, tần suất người mang gen bệnh trong các quần thể không đồng đều: ở Nhật Bản: 1/65; Trung Quốc: 1/63; Hàn Quốc: 1/108. Hiện chưa có nghiên cứu nào về tần suất người mang gen bệnh tại Việt Nam.
Triệu chứng lâm sàng:
Thiếu hụt Citrin được chia thành hai thể lâm sàng dựa vào độ tuổi khởi phát:
- Viêm gan ứ mật ở sơ sinh (Neonatal intrahepatic cholestatic hepatitis caused by citrin deficiency – NICCD)
- Tăng citrullin typ II khởi phát ở tuổi trưởng thành (Adult-onset type II Citrullinemia – CTLN2).
Ngoại trừ một số ca bị rối loạn chức năng gan nghiêm trọng cần phải ghép gan, NICCD thường không nghiêm trọng vì các triệu chứng sẽ biến mất khi trẻ đầy 1 tuổi. Tuy nhiên, sau 10 năm hoặc lâu hơn nữa, một số bệnh nhân có thể chuyển sang CTLN2 với các triệu chứng liên quan đến thần kinh, có thể nguy hiểm đến tính mạng Ngoài ra, các triệu chứng, và các kỹ thuật sử dụng trong chẩn đoán xác định bệnh thiếu hụt Citrin như xét nghiệm sinh hóa thường không đặc hiệu. Hiện nay, kỹ thuật sinh học phân tử là phương pháp duy nhất để chẩn đoán xác định bệnh nhân nghi ngờ mắc thiếu hụt Citrin, góp phần quan trọng trong phát hiện sớm và ngăn chặn NICCD chuyển sang CTLN2.
- NICCD: Xuất hiện ở trẻ sơ sinh với vàng da kéo dài, rối loạn chức năng gan với viêm mật, viêm gan. Từ 0-6 tháng tuổi: axit amin tăng cao bao gồm citrullin, threonin, methionin, tyrosin, phenylalanine, arginin; bilirubin, tăng axit mật tổng số (Total bile axit-TBA), tăng α – fetoprotein; galactosemia; hypoproteinemia; hypoglycemia.
- CTLN2: Khởi phát một cách đột ngột ở tuổi trưởng thành. Xuất hiện các triệu chứng thần kinh bao gồm mất phương hướng, tập tính bất thường: gây gổ, cáu bẳn, tăng động, hôn mê, và có thể tử vong do hôn mê não. Thích ăn thực phẩm giàu protein, không thích ăn thực phẩm giàu cacbonhydrat như Cơ thể mảnh khảnh, rối loạn chức năng gan: viêm gan, viêm mật. Tăng citrullin, amoniac máu, PSTI (Pancreatic secretory trypsin inhibitor). Giảm ASS (Arginosuccinate synthetase); giảm tỉ lệ Fischer (tỉ lệ Val+Leu+Ile/Tyr+Phe).
Cơ sở phân tử:
Gen gây bệnh của cả hai thể lâm sàng nằm trên cánh dài của nhiễm sắc thể số 7 (7q21.3), có kích thước khoảng 200 kb, gồm 18 exon và mã hóa cho 675 axit amin (aa), cấu tạo thành Citrin. Citrin chủ yếu biểu hiện ở gan là cơ quan đóng vai trò chuyển hóa nhiều chất khác nhau. Nhiều đột biến đã được tìm thấy trên gen SLC25A13, tuy nhiên, chỉ một số đột biến thường gặp: đột biến số I (851del4), đột biến số III (1638ins23), đột biến số X (IVS6+5 G→A) và đột biến số XIX (IVS16ins3kb).
Cơ chế di truyền:
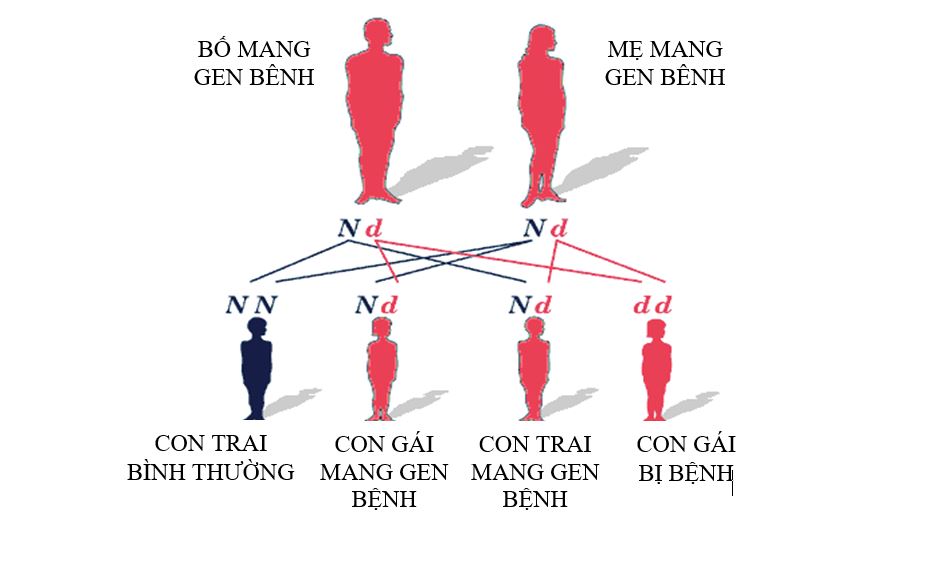
Thiếu hụt Citrin là bệnh di truyền lặn trên nhiễm sắc thể thường. Tỷ lệ mắc bệnh là như nhau ở cả hai giới nam và nữ.
Bố và mẹ là mang gen dị hợp tử với một đột biến, nguy cơ trong một lần sinh như sau:
- 50% số con là người lành mang gen bệnh.
- 25% số con là người hoàn toàn khỏe mạnh không mang gen bệnh
- 25% số con mắc bệnh thiếu hụt Citrin.
Chẩn đoán xác định:
- Xét nghiệm gen SLC25A13
Phương pháp xét nghiệm: Phân tích 4 đột biến trên gen SLC25A13.
- Tách chiết ADN
- Kĩ thuật PCR: Để xác định đột biến trên gen SLC25A13, ADN genome của mỗi bệnh nhân được dùng làm khuôn cho phản ứng PCR nhờ các cặp mồi đặc hiệu đã được thiết kế sẵn.
- PCR-RFLP: Dựa vào vị trí nhận biết của enzyme giới hạn, sản phẩm PCR của đột biến số I, X, XIX sẽ được cắt enzyme ở điều kiện thích hợp: đột biến số I, số X lần lượt được nhận biết bằng cắt enzyme
- Điện di: Sản phẩm PCR của đột biến số III và sản phẩm cắt enzyme của các đột biến còn lại được điện di trên thạch agarose 2% để nhận biết kích thước.
Tài liệu tham khảo:
Kobayashi K, Saheki T (2003) Aspartate glutamate carrier (citrin) deficiency. In: Broer S and Wagner CS (eds) Membrane Transporter Diseases. Kluwer Academic/Plenum Publishers, New York, pp 147-60.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999; 22: 159–63.
Kobayashi K, Lu YB, Li MX, Nishi I, Hsiao KJ, Choeh K, Yang Y, Hwu WL, Reichardt JKV, Palmieri F, Okano Y, Saheki T. Screening of nine SLC25A13 mutations: their frequency in patiens with citrin deficiency and high carrier rates in Asian populations. Molecular Genetics and Metabolism. 2003; 80 pp 356-359.
Kobayashi K, Ushikai M, Song Y-Z, Sheng J-S, Tabata A, Gao H-G, Okumura F, Saheki T. Overview of citrin deficiency: SLC25A13 mutations and the frequency. J Appl Clin Pediatr.