Định nghĩa:
Chẩn đoán bệnh X-linked Lymphoproliferative (XLP) nên được đặt ra ở những trường hợp người nam với biểu hiện nhiễm cấp Epstein-Barr virus (EBV), làm tăng bạch cầu đơn nhân trung tính, có thể kèm theo hiện tượng thực bào tế bào máu. Ở các trường hợp này, nếu bệnh nhân vượt qua được giai đoạn nhiễm EBV tiên phát, có sự biểu hiện của suy giảm chỉ số gammaglobulin máu ở các mức độ khác nhau, và có nguy cơ cao với việc phát triển bệnh thành u lumpho ác tính hoặc các nhóm bệnh có tăng sinh tế bào lympho khác. Các triệu chứng của bệnh XLP đôi khi cũng xuất hiện trên một số bệnh nhân không nhiễm EBV.
Cơ sở phân tử:
Có hai gen liên quan đặc hiệu đến hội chứng XLP.
SH2D1A, là một trong hai gen gây nên bệnh XLP, nằm trên cánh dài nhiễm sắc thể số 6 (Xq24-26). Đột biến trên gen SH2D1A tìm thấy ở khoảng 60% trường hợp XLP. Gen SH2D1A mã hóa cho 128 amino acid, và cho loại protein có tên là SAP (SLAM associated protein), tham gia vào tín hiệu hoạt động của tế bào lympho. Gen SH2D1A bao gồm 4 exon và 3 intron. Đột biến trên gen SH2D1A đã được tìm thấy trên tất cả 4 exon, trong đó đột biến mất đoạn lớn chiếm khoảng 25%. Chức năng của tế bào NK và biểu hiện protein SAP được đánh giá bằng flowcytometrie luôn biểu hiện bất thường trên các bệnh nhân có đột biến gen SH2D1A.
BIRC4, là gen thứ hai gây XLP, nằm trên nhánh dài nhiễm sắc thể X (Xq25). Đột biến trên gen BIRC4 được tìm thấy trên khoảng 20% bệnh nhân mắc XLP. BIRC4 mã hóa 497 amino acid, và cho protein có tên XIAP ( X-linked inhibitor of apoptosis). Đột biến trên gen BIRC4 dẫn đến sự khiếm khuyết của quá trình apoptosis. Gen BIRC4 bao gồm 6 exon và 5 intron. Mất đoạn lớn cũng là một dạng đột biến được tìm thấy trên gen BIRC4. Chức năng tế bào NK luôn giảm ở những bệnh nhân có đột biến gen BIRC4, trong khi đó biểu hiện protein SAP có thể bình thường. Hiện nay, chưa có phương pháp nào đặc hiệu để đánh giá biểu hiện protein XIAP. Lách to là một triệu chứng đựoc ghi nhận ởm ột vài bệnh nhân có đột biến gen BIRC4, tuy nhiên đây không phải là triệu chứng đặc hiệu để phân biệt XLP thứ phát và XLP có đột biến trên gen SH2D1A.
Cơ chế di truyền:
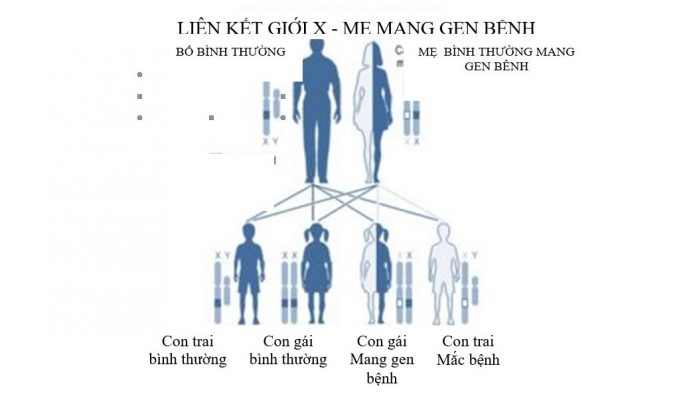
Bệnh XLP là bệnh di truyền liên kết nhiễm sắc thể giới tính X. Mẹ là người bình thường mang gen bệnh, di truyền bệnh cho con trai. Trong một lần sinh, nguy cơ của những người mẹ này như sau:
- 50% số con gái là người bình thường mang gene bệnh.
- 50% số con gái là người bình thường không mang gen bệnh
- 50% số con trai là người bình thường không mang gen bệnh
- 50% số con trai là người mắc bệnh XLP
Chẩn đoán:
- Triệu chứng lâm sàng của từng bệnh nhân.
- Yếu tố nguy cơ
- Phát hiện người nữ mang gen bệnh trong tiền sử gia đình.
Phương pháp xét nghiệm: Phân tích gen SH2D1A và BIRC4:
- Tách chiết DNA
- Kỹ thuật PCR: để xác định đột biến trên gen SH2D1A và BIRC4, ADN genome của mỗi bệnh nhân được dùng làm khuôn cho phản ứng PCR nhờ các cặp mồi đặc hiệu đã được thiết kế sẵn.
- Giải trình tự gen trực tiếp cho toàn bộ vùng exon, intron và vùng giáp ranh intron/exon của SH2D1A và Ở người nam, giải trình tự gen có thể phát hiện khoảng 98% đột biến trên gen SH2D1A. Ở người nữ, phương pháp này có thể phát hiện khoảng 75% đột biến trên gen SH2D1A, mất đoạn lớn gặp ở khoảng 25% đột biến.
- Biểu hiện của protein SAP được đánh giá bằng flow cytometry có thể hiệu quả để định hướng chẩn đoán và xác định gen cần phân tích.
Tài liệu tham khảo:
Ueda I, Morimoto A, Inaba T, Yagi T, Hibi S, Sugimoto T, Sako M. Characteristic perforin gene mutations of haemaphagocytic lymphohistiocytosis patients in Japan. Br J Hematol 2003; 121:503-10.
Henter JI, Samuelsson-Home A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S. Treatement of hemophagocytic lymphohistiocytosis with HLH-94 mmunochemotherapy and bone marrow transplantation. Blood 2002;100:2367-73.
Henter JI, Arico M, Elinmder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12:417-33.