Định nghĩa:
Tăng sản thượng thận bẩm sinh (TSTTBS) là bệnh di truyền lặn trên nhiễm sắc thể thường, được đặc trưng bởi sự thiếu hụt hoạt động của một trong các enzyme cần thiết để tổng hợp cortisol từ cholesterol của vỏ thượng thận như: 21-hydroxylase (21-OH), 11β-hydroxylase (11-OH), 17- hydroxylase (17-OH), 3β- hydroxysteroid dehydrogenase (3β- HSD) và 20/22 Desmolase. Trong đó, 90- 95% trường hợp TSTTBS là do thiếu hụt hoạt động của enzyme 21-OH.
Triệu chứng lâm sàng:
Sự thiếu hụt hoạt động của 21-OH dẫn đến sự thiếu hụt cortisol và/ hoặc thiếu hụt aldosterone cũng như thừa adrogen thượng thận. Biểu hiện lâm sàng của TSTTBS tuỳ thuộc vào mức độ thiếu hụt hoạt động của enzyme, bao gồm:
- Thể nam hoá đơn thuần: có biểu hiện nam hoá bộ phận sinh dục ngoài ở trẻ gái và tình trạng giả dạy thì sớm ở trẻ trai.
- Thể mất muối: với các triệu chứng lâm sàng tương tự, và có kèm theo thếu hụt aldosterone ở trẻ sơ sinh.
- Thể không cổ điển hoặc khởi phát muộn: với các triệu chứng lâm sàng như giả dậy thì sớm ở trẻ gái, hội chứng rậm lông, buồng trứng đa nang và giảm khả năng thụ tinh.
Cơ sở phân tử:
Vùng gene mã hoá cho enzyme 21-OH gồm 2 gene: gene CYP21 và gene giả CYP21P- nằm giữa vùng phức hợp hoà hợp mô chủ yếu (Major Histiocompatibility Complex- MAC- cạnh gene HLA). Vùng gene này nằm trên cánh ngắn của nhiễm sắc thể số 6 (6p2.1-3) có độ lớn xấp xỉ 30kb. Gene CYP21 và CYP21P nằm xen kẽ với hai gene mã hoá cho bổ thể C4A và C4B. Gene CYP21 là thành viên của nhóm P450 mà đặc hiệu bởi C21 trong tiền chất steroid vỏ thượng thận. Gene CYP21P là gene bất hoạt, gene CYP21 mã hoá cho enzyme 21-OH. Mỗi gene CYP21 và CYP21P có độ lớn xấp xỉ 3kb gồm 10 exon, hai gene này có trình tự tương đồng giống nhau đến 98%. Trình tự của giả gene CYP21P có các loại đột biến thường thấy trên bệnh nhân TSTTBS. Qua quá trình phân bào giảm nhiễm, sự bắt chéo không đồng đều giữa các cặp nhiễm sắc thể sẽ dẫn tới việc các trình tự ở vùng giả gene sẽ chuyển sang gene CYP21 gây nên đột biến trên gene. 95% các đột biến trên gene CYP21 là do đột biến chuyển đoạn từ giả gene CYP21P, 5% còn lại là do tự gene CYP21 bị đột biến. Kiểu đột biến có liên quan chặt chẽ đến kiểu hình của bệnh TSTTBS.
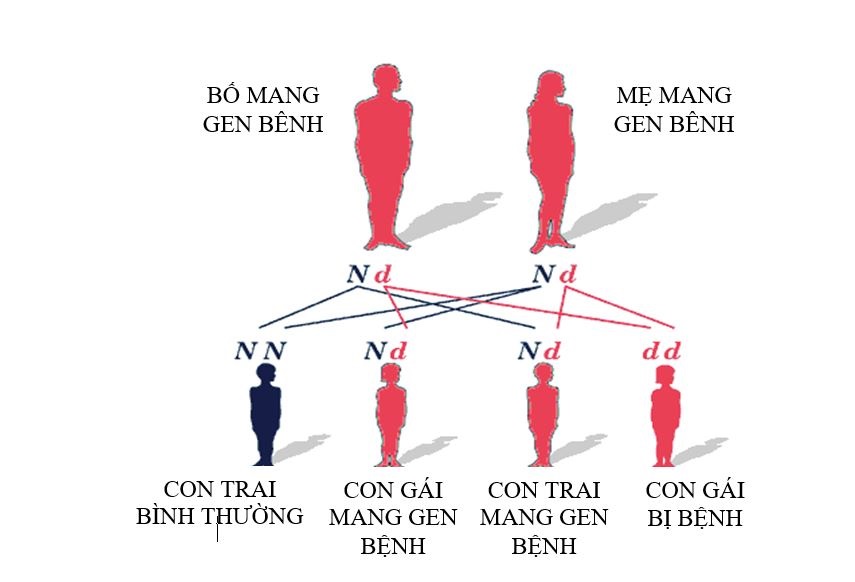
Cơ chế di truyền:
TSTTBS là bệnh di truyền lặn trên NST thường. Tỷ lệ mắc bệnh là như nhau ở cả hai giới nam và nữ.
Bố và mẹ là mang gen dị hợp tử với một đột biến, nguy cơ trong một lần sinh như sau:
- 50% số con là người lành mang gen bệnh.
- 25% số con là người hoàn toàn khỏe mạnh không mang gen bệnh
- 25% số con mắc bệnh TSTTBS.
Phương pháp xét nghiệm: Phân tích gene CYP21
- Chiết tách DNA
- Sàng lọc các đột biến thường gặp bằng kỹ thuật MLPA: Sử dụng kit MLPA P050B2 (MRC- Holland) sàng lọc 06 đột biến thường gặp: P30L (Exon 1), Mất đoạn 8bp (Exon 3), I172N (Exon 4), Exon 6 Cluster (Exon 6), Q318 (Exon 8), Mất toàn bộ gene CYP21, Mất đoạn 30Kb (Exon 3 hybrid). Kết quả được phân tích trên máy CEQ8000 (Beckman Coulter).
- Giải trình tự gene CYP21: Để phát hiện các đột biến khác
Tài liệu tham khảo:
Keegan CE, Killeen AA. 2001. An overview of molecular diagnosis of steroid 21- hydroxylase deficiency. JMD. 3(2).
New MI. 2001. Prenatal treatment of congenital adrenal hyperplasia, the United States experience. Endocrinology Metab Clin North Am. 30(1): 1-13.
New MI, Carslson A, et al. 2003. Update: Prenatal diagnosis for congenital adrenal hyperplasia in 595 pregnancies. The Endocrinologist. 13(3): 233-39.
Perrin C, White et al. 2000. Congenital adrenal hyperplasia due to 21- hydroxylase deficiency. Endocrine Reviews. 21: 245- 91.
Speicer PW, Laforgia N et al. 2003. Congenital adrenal hyperplasia. N Engl J Med. 349(8): 76- 89.